





应用领域
技术路线
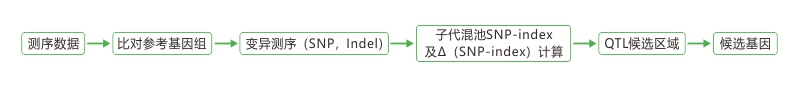
分析内容
|
1.测序数据质量评估 (碱基组成分布等)
2.原始数据过滤,去除含接头reads、低质量reads
3.与参考基因组比对
3.1 比对基因组统计
3.2 插入片段统计
3.3 基因覆盖度分析
3.4 测序深度分析
4.SNP分析统计
4.1 SNP 检测
4.2 SNP位置和功能注释
4.3 SNP转换和颠换信息统计
4.4 SNP杂合信息统计
|
5.InDel 分析统计
5.1 InDel 检测
5.2 InDel 位置和功能注释
5.3 InDel杂合信息统计
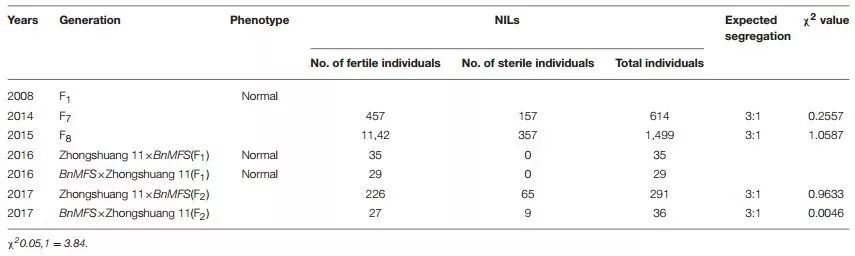
6. BSA分析
6.1 亲本间多态性比较
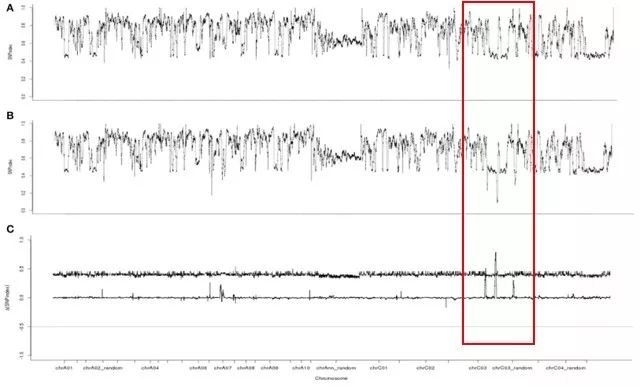
6.2子代SNP频数分析(SNP index和 ΔSNP index计算)
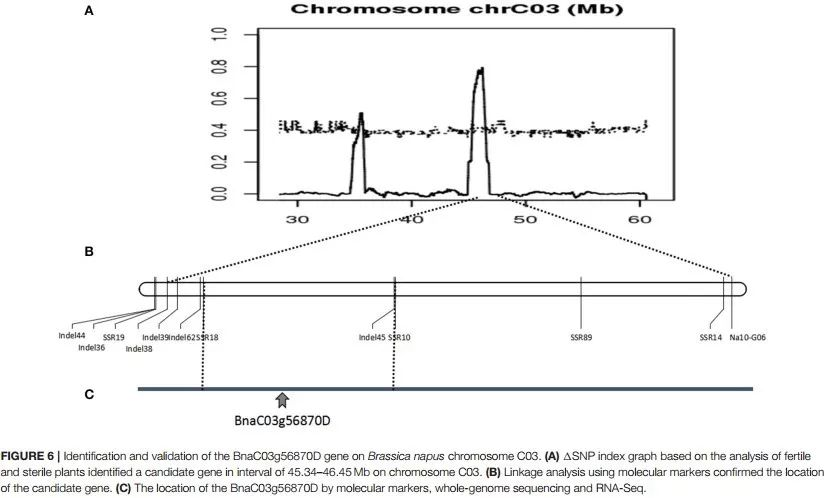
6.3 目标性状相关区域定位
6.4 目标区域候选基因分析
|
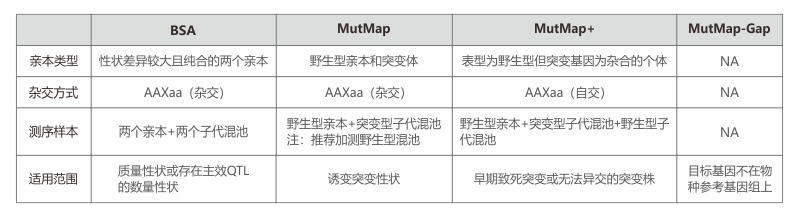
BSA + RNA-seq:高效筛选目标基因
研究样本
研究思路
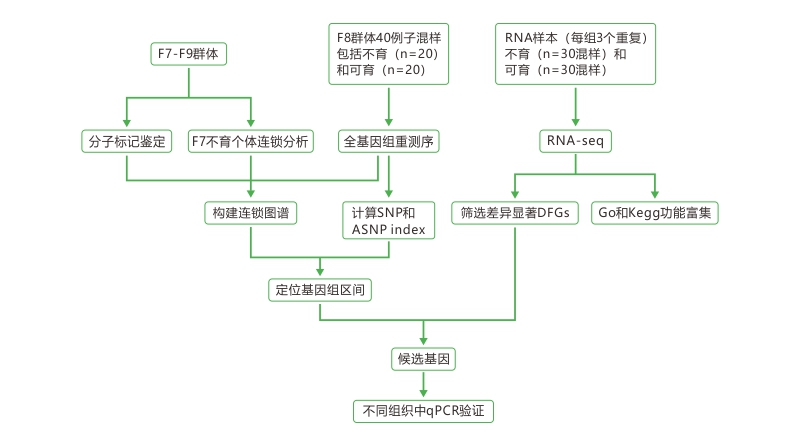
研究结果