












|
基因组重测序是在已知物种基因组的情况下进行测序。通过对该物种的不同个体或同一个体的不同组织进行基因组重测序,可从群体或个体水平全面挖掘基因组序列差异。该方法能以较低的成本获得大量、准确的基因组信息,对动植物育种研究及人类疾病等方面具有重大的指导意义。 |
应用领域
|
1. 个体基因组变异检测
2. 突变体功能突变位点定位
3. 群体遗传学分析
4. 全基因关联分析
|
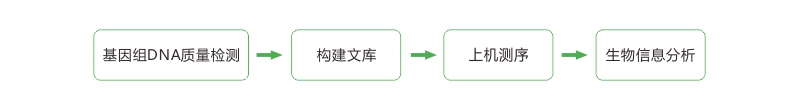
技术路线
分析内容
|
1. 标准分析:原始数据基本分析、序列比对、SNP检测等;
2. 高级分析:InDel检测、CNV检测、SV检测等;
3. 个性化分析:群体遗传学分析、GWAS分析等。
|
样品要求
|
样品类型:无降解或者轻微降解、无RNA污染的DNA样品; 样品需求量:≥1 μg;样品浓度:>20 ng/μL;样品纯度:OD260/280= 1.8~2.0。 |
项目周期
| 一般在50个工作日,具体完成时间视项目具体规模而定。 |
|
参考文献 [1] Ryan E. Mills, Klaudia Walter, Chip Stewart, Robert E. Handsaker, et al. Mapping copy number variation by population-scale genome sequencing. Nature February 2011 470.59–65
[2] Srikanth Gottipati, Leonardo Arbiza, Adam Siepel, Adam Siepel, Andrew G Clark, Alon Keinan. Analyses of X-linked and autosomal genetic variation in population-scale whole genome sequencing. Nature Genetics 43 741–743 (2011)
[3] Can Alkan , Bradley P. Coe & Evan E. Eichler Genome structural variation discovery and genotyping Nat. Rev. Genet., 12, 363–376 (2011).
[4] Derek M. Bickhart, Yali Hou, Steven G. Schroeder, et al. Copy number variation of individual cattle genomes using next-generation sequencing February 2, 2012, doi: 10.1101/gr.133967.111
[5] Kerstin Lindblad-Toh, Manuel Garber, Or Zuk,Michael F. Lin, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482(2011)
[6] Michael F. Berger, Eran Hodis, Timothy P. Heffernan, Yonathan Lissanu Deribe, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 11071 (2012)
[7] Tobias Rausch, David T.W. Jones, Marc Zapatka, Adrian M. Stütz, Thomas Zichner, Joachim et al. Genome Sequencing of Pediatric Medulloblastoma Links Catastrophic DNA Rearrangements with TP53 Mutations. Cell, Volume 148, Issue 1, 59-71, 20 January 2012
[8] Zamin Iqbal, Mario Caccamo,Isaac Turner, Paul Flicek, Gil McVean De novo assembly and genotyping of variants using colored de Bruijn graphs. Nature Genetics 44, 226–232 (2012)
|
|
Q1: 可以用简化基因组测序来做QTL-seq分析吗? A:由于简化基因组技术(GBS/RAD)是对基因组上的酶切片段进行测序,数据量一般只有基因组的1~10%,有可能会降低QTL定位的精度。同时,也无法进一步筛查QTL区间内的突变信息。因此,我们建议用全基因组重测序来进行QTL-seq分析。
Q2: 群体进化分析,需要选择简化基因组测序还是全基因组重测序? A:考虑到自然群体极快的LD衰减距离, 简化基因组的覆盖度显得有所不足,全基因组重测序是趋势。推荐样本的全基因组重测序深度要大于基因组的10x。
Q3: 怎么确定性状是质量性状还是数量性状? A:根据统计子代个体的性状分离比进行判断。质量性状子代分离比例为3:1,主效基因控制的数量性状子代分离比例接近3:1。
|
全基因组重测序揭示高粱未开发的遗传潜力
| 发表期刊《Nature communications》 |
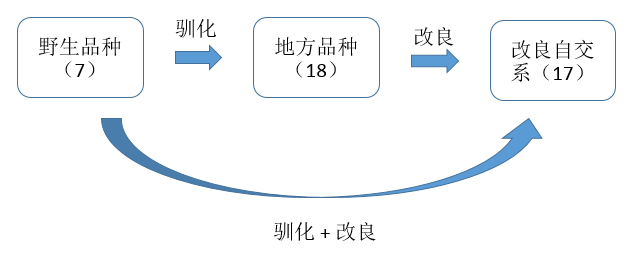
研究对象:
| 44株不同来源的高粱(包括7株野生品种、18株地方品种、17株改良自交品种、2株来自西非的地方品种)、和2株近缘种(S. propinquum,拟高梁)。 |
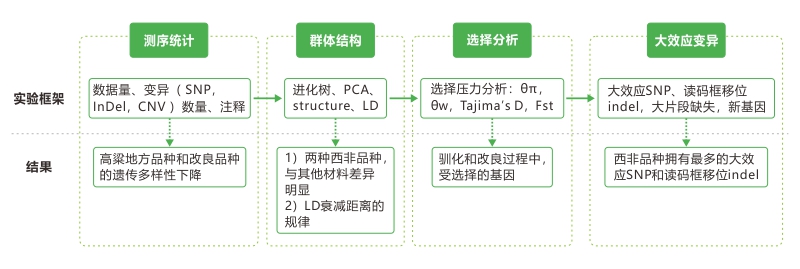
研究思路:
研究结果
一. 测序数据和变异统计
|
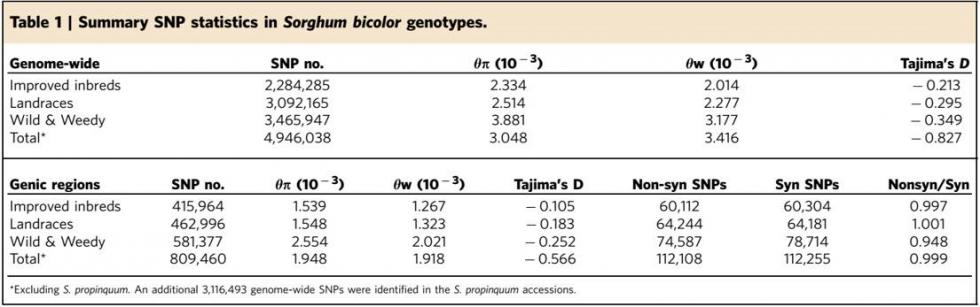
1. 文章鉴定出了大量的SNP, InDel和CNV,这些变异绝大部分位于基因间区,编码区则相对比较保守。 2. 野生品种比地方品种和改良品种有更多、密度更高的SNP,改良品种的SNP数目和indel数目最少。西非品种Guinea-margaritiferum的SNP密度是地方品种的两倍。 另外,θπ和θw检验结果也表明了改良品种的基因多样性最低。这些结果表明遗传多样性在高粱驯化和改良的过程中不断下降,并且发生了遗传瓶颈效应。同时也提示了野生高粱种质具有很大的开发和改良潜力。 |
 |
二. 群体结构和种群历史分析
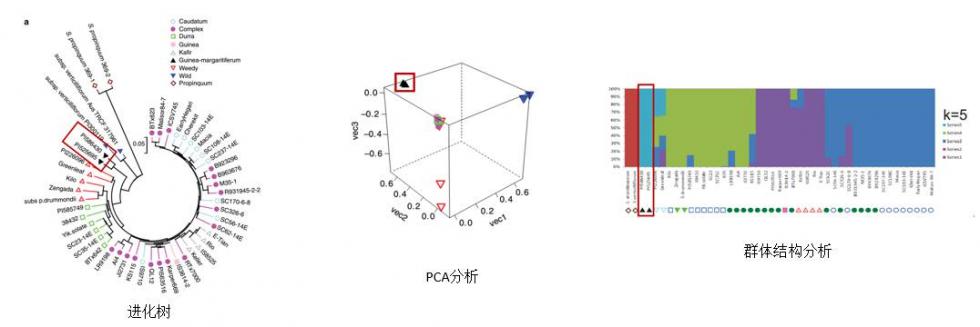 |
进化树结果显示,西非品种Guinea-margaritiferum处于栽培种和野生种之间;PCA分析和群体结构分析结果表明Guinea-margaritiferum离群。不同的研究方法都揭示了相同的结论,即高粱品种之间强烈的种群结构差异和复杂的驯化历程,并证实了Guinea-margaritiferum是发生在西非的第二次独立驯化事件所产生的品种。 |
三. 全基因组扫描筛选受驯化选择基因
|
结果总共找到725个与驯化和改良相关的候选基因,其中33%(285个)的驯化候选基因呈现出改良历程中的受选择信号,预示着这些基因在农艺性状改良上有着重要的作用。 对这些候选基因进行基因功能注释,发现功能富集于生物胁迫和非生物胁迫引起的植物激素响应,例如GH3,SAUR, IAA基因家族。这些驯化与改良候选基因为鉴定与农艺性状相关的基因提供了宝贵的资源。 通过这种选择压力分析得到的这些候选基因涉及的性状多种多样,涉及的基因功能也多种多样,如:dep1 :产量提高相关基因(水稻);Psy1:控制种子颜色(玉米);Bif1:株型(玉米)还有很多与驯化农艺性状相关的基因,如种子相关性状、植株颜色、株高等。 |
 |
|
参考文献: Mace, Emma S., et al. "Whole-genome sequencing reveals untapped genetic potential in Africa’s indigenous cereal crop sorghum." Nature communications 4.1 (2013): 1-9. |











