外显子测序揭示纳米调控肿瘤细胞DNA损伤以增强基于新抗原的胰腺癌免疫治疗
|
合作单位:中国科学院大学温州研究所
发表期刊:Biomaterials
|
|
目的:肿瘤免疫治疗中,低免疫原性肿瘤对免疫治疗的反应率低,疗效不佳。这类肿瘤的肿瘤突变负荷(TMB)较低,导致肿瘤抗原或新抗原表达不足,免疫原性差。本研究开发了一种基于金属有机框架(MOFs)的纳米调节器,用于同时递送DNA损伤剂和DNA损伤修复抑制剂,通过干预肿瘤DNA稳定性,增加肿瘤突变负荷,从而提高肿瘤免疫原性,有望提高低免疫原性肿瘤免疫治疗的疗效。
取材:经 MOFDOX&siATR 处理的 Panc02 细胞作为实验组,以及未经处理的 Panc02 细胞作为对照组。收获用纳米调节剂处理的肿瘤细胞用于 DNA 提取和随后的基因组分析
结果:本文通过细胞实验和动物实验验证了MOFDOX&siATR纳米调节器在提高低免疫原性胰腺癌免疫原性方面的有效性。结果表明,MOFDOX&siATR能够有效诱导肿瘤细胞DNA损伤,抑制DNA损伤修复,增加肿瘤突变负荷,并促进新抗原的产生。此外,MOFDOX&siATR还能够诱导肿瘤细胞发生免疫原性细胞死亡,并促进树突状细胞(DC)的成熟和活化,从而增强抗肿瘤免疫反应。在动物实验中,MOFDOX&siATR显著抑制了肿瘤生长,并诱导了抗肿瘤免疫记忆,表现出良好的抗肿瘤疗效。
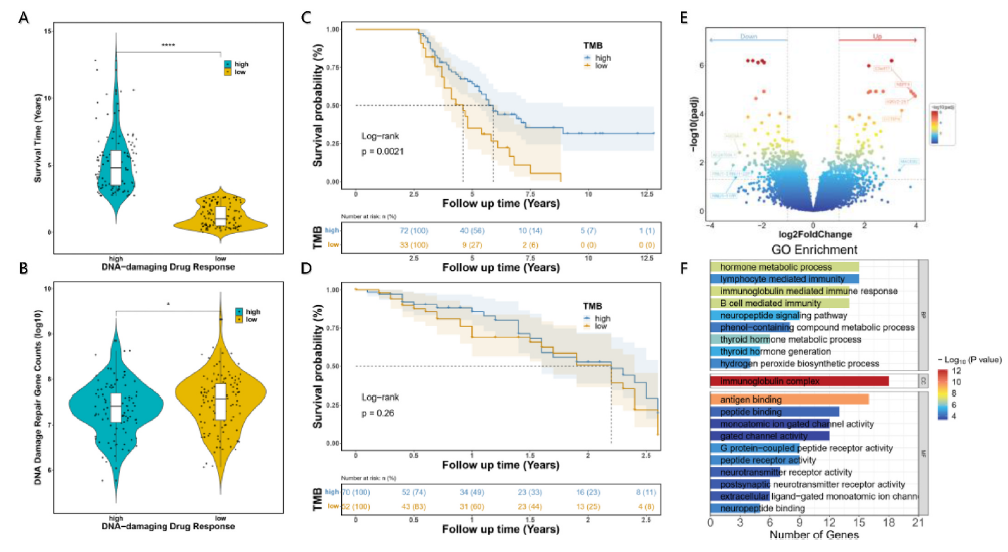
图1 临床低免疫原性肿瘤治疗的生物信息学分析
参考文献
Wang J, Wu C, Wang Y, et al. Nano-enabled regulation of DNA damage in tumor cells to enhance neoantigen-based pancreatic cancer immunotherapy[J]. Biomaterials, 2024, 311: 122710.
|