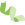
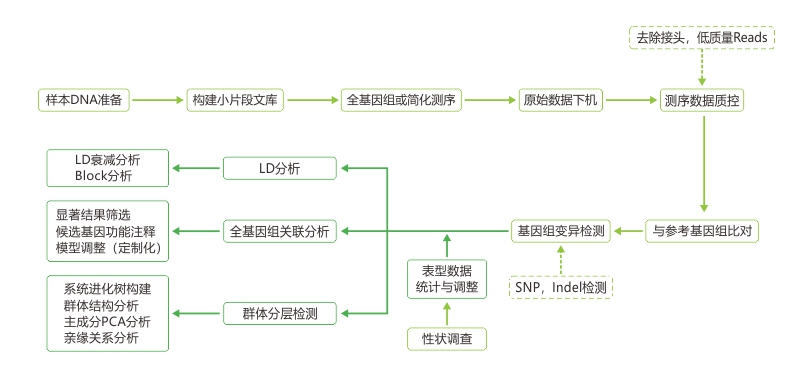
|
参考文献
[1] Lin H, Wang F, Rosato AJ, Farrer LA, Henderson DC, Zhang H. Prefrontal cortex eQTLs/mQTLs enriched in genetic variants associated with alcohol use disorder and other diseases [published online ahead of print, 2020 Jun 4]. Epigenomics. 2020;10.2217/epi-2019-0270.
[2] Loh NY, Minchin JEN, Pinnick KE, et al. RSPO3 impacts body fat distribution and regulates adipose cell biology in vitro. Nat Commun. 2020;11(1):2797. Published 2020 Jun 3.
[3] Maroteau C, Kalhan Siddiqui M, Veluchamy A, et al. Exome sequencing reveals common and rare variants in F5 associated with ACE inhibitor and ARB induced angioedema [published online ahead of print, 2020 Jun 4]. Clin Pharmacol Ther. 2020;10.1002/cpt.1927. doi:10.1002/cpt.1927.
[4] Howell AE, Robinson JW, Wootton RE, et al. Testing for causality between systematically identified risk factors and glioma: a Mendelian randomization study. BMC Cancer. 2020;20(1):508. Published 2020 Jun 3. doi:10.1186/s12885-020-06967-2
|