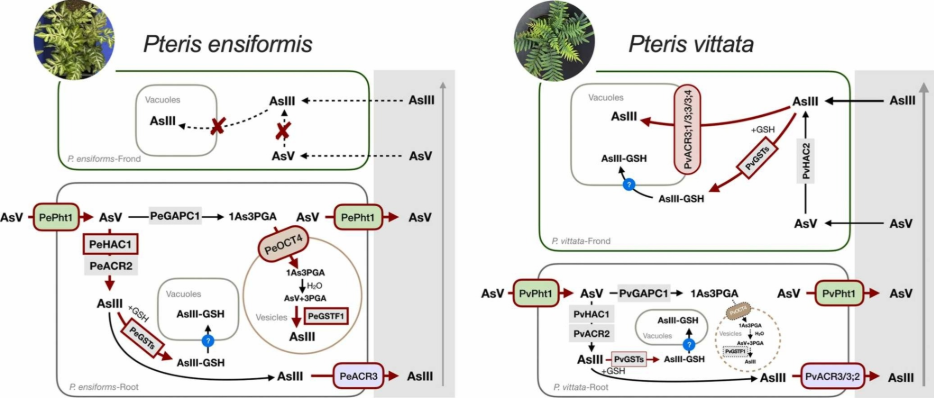






应用领域
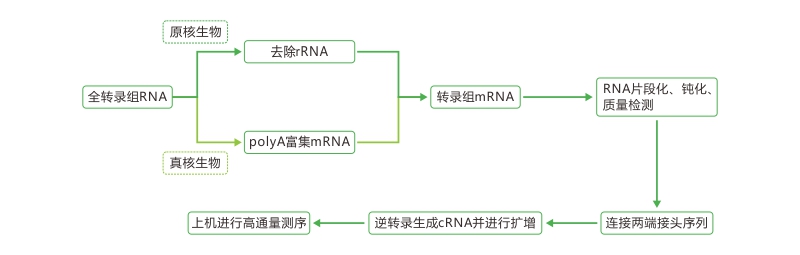
技术路线
分析内容
标准信息分析
|
1. 原始数据过滤,去除接头序列及低质量reads 2. 数据产出统计及测序质量评估(测序饱和度分析、测序随机性分析) 3. 转录组denovo组装 4. 组装质量统计与分析(Contig N50、Unigene*长度分布、reads覆盖统计) 5. Unigene表达统计(基因覆盖度,表达量、表达量丰度分布) 6. Unigene基本功能注释 6.1 Unigene Nr蛋白数据库注释 6.2 Unigene SwissProt蛋白数据库注释 6.3 Unigene COG/KOG注释及分类 6.4 Unigene GO功能注释及分类 6.5 Unigene Pathway代谢通路注释 7. Unigene高级功能注释 7.1 预测编码蛋白框(CDS)及Unigene序列方向预测 *此处Unigene 等同于“转录本”或“Transcript” 7.2 近缘模式生物同源基因CDS比较 7.3 Pfam蛋白结构域分析 |
8. SSR开发及引物设计 9. 样本关系分析(主成分分析(PCA)、相关性系数热图、样本聚类图) 10. 差异表达基因分析(两个或两个以上样品) 10.1 差异表达基因筛选
10.2 差异基因火山图
10.3 差异基因表达模式聚类分析(热图)
10.4 差异基因GO功能显著性富集分析
10.5 差异基因Pathway显著性富集分析
11 互作网络分析 12 GSEA分析 13 突变分析(SNP/InDel分析) 14 omicmart 在线报告
|
高级信息分析